
Ausgewählte Tumore im Kindesalter
Unter solidem Tumor versteht man grundsätzlich eine Geschwulst, die gutartig oder bösartig sein kann. Bei Kindern sind Tumore und hier vor allem die bösartigen grundsätzlich anders als diejenigen des Erwachsenen. Erwachsene erkranken unter Prostata-, Brust-, Bronchus- und Dickdarmkarzinomen, Kinder entwickeln beispielsweise ein Neuroblastom (leitet sich von Nervenzellen – sympathische Ganglien - ab), ein Nephroblastom oder auch Wilms – Tumor (Mischtumor aus fehldifferenziertem embryonalen Nierengewebe), ein Teratom (oftmals in der Steißbeinspitze beginnender kongenitaler Tumor aus Geweben aller drei Keimblätter) oder schon viel seltener Gonadale Tumoren (Eierstock- bzw. Hodentumoren). Pro Million Einwohner ist jährlich mit etwa 50 Neuerkrankungen an Leukämie und Lymphomen, 20 Tumoren des zentralen Nervensystems und 30 soliden Tumoren (vorwiegend Neuroblastome, Wilmstumore und Sarkome) zu rechnen. Die, gegenüber dem Erwachsenen – Krebsgeschehen andersartigen bösartigen Tumoren des Kindesalters ermöglichen grundsätzlich effektivere Heilungschancen. Diese Chancen werden in mulimodalen Therapiekonzepten vereinigt. Multimodal bedeutet, dass eine interne Therapie (Chemotherapie, Knochenmarkstransplantation) durch eine kinderchirurgische und gegebenenfalls radiologische Therapie (Strahlentherapie) ergänzt wird. Das beinhaltet die intensive Zusammenarbeit des Ärzteteams wo als wesentliche Komponente die Angehörigen eingebunden werden müssen.
Tumore im Kindes- und Jugendalter sind charakterisiert durch bestimmte Altersgipfel, Geschlechtsgebundenheit sowie fallweise eine genetische Prädisposition. Manche Tumore haben einen Häufigkeitsgipfel bereits in den ersten Lebensmonaten wie z.B. das Retinoblastom und Neuroblastom, Leukämien zwischen 2-5 Jahren, Wilmstumore und Neuroblastome zwischen 2-5 Jahren, Ewing- und Osteosarkome zwischen 10-18 Jahren. Die meisten kindlichen Tumore treten bevorzugt beim männlichen Geschlecht auf; für manche, wie das Ewing-Sarkom und das Rhabdomyosarkom wird dies erst im höheren Schulalter deutlich. Teratome sind häufiger bei Mädchen (75%), allerdings ist die maligne Potenz bei Knaben höher.
Kombinationen mit angeborenen Fehlbildungen sind bekannt: z.B. Kinder mit Aniridie erkranken 1.000x häufiger an Wilmstumor. Hemihypertrophie, Beckwith-Wiedemann-Syndrom, Hamartome können mit Wilms-Tumor, Nebennierentumoren oder Lebertumoren kombiniert sein. Eine hereditäre Belastung ist beim Retinoblastom erwiesen, ebenso die Kombination mit Trisomie 21. Ein hohes Krebsrisiko haben primäre Lymphangiome, Wiskott-Aldrich Syndrom, Ataxie-Teleangiektase Syndrom, M. Recklinghausen, Immundefizitsyndrome und Immundepression nach Transplantation. Genetisch virale Interaktionen werden z.B. bei B-Zell Lymphom, Karposi-Sarkom, hepatozellulärem Karzinom und nasopharyngealem Karzinom angenommen. Die Wichtigkeit der Gene und Erblichkeit gewinnt gerade bei kindlichen Tumoren zunehmend an Bedeutung. So scheinen 40% der Retinoblastome erblich bedingt zu sein (Retinoblastoma locus am Chromosom 13, ähnliche Onkogene sind für Patienten mit Wilmstumor, Osteosarkom, Hepatoblastom und Rhabdomyosarkom entdeckt worden. Kinder mit Trisomie 21 haben eine 10-fache Häufigkeit für akute lymphatische Leukämie. Viele Translokationen von Chromosomen spezifisch für bestimmte Tumore sind identifiziert worden (z.B. Translokation 11/22 beim PNET), einige Onkogene sind tumorspezifisch (z.B. N-myc für Neuroblastom). Manche Tumore haben interessante biologische Eigenschaften der Spontanregression, am besten bekannt beim Neuroblastom.
Neuroblastom
Neuroblastome entstehen aus sympathischen Ganglien oder dem Nebennierenmark. Dementsprechend finden sich diese Geschwülste nahe dem Rückenmark und speziell von den Nebennieren ausgehend. Die Tumore schütten „Stresshormone“ aus (Adrenalin, Noradrenalin) und dementsprechend sind die Hormanabbauprodukte im Harn (beispielsweise Vanilinmandelsäure – Bestimmun im 24h Harn) erhöht. Die Symptome sind meist nicht sehr eindrucksvoll, bestehen aus Durchfällen, einer Anämie neurologischen Ausfällen, Knochenschmerzen und einem vorgewölbten Abdomen, letzteres erst in fortgeschrittenen Stadien. Die Stadien gehen von I – IV (IVs). Das IV s Stadium beschreibt einen Tumor in der Größe eines Stadium I – II, allerdings beispielsweise mit Knochenmarksbefall. Die Diagnostik ergibt sich aus einem typischen CT. Metastasen sind in Lymphknoten, Leber und Lunge möglich. Wird der Tumor vor dem ersten Lebensjahr diagnostiziert und trägt Merkmale einer günstigen Tumorbiologie (n-myc Amplifikation neg.; hyperdiploider Chromosomensatz; CD 44 Expression) so können auch Selbstheilungen (Ausreifen des Tumors zu einem Ganglion) vorkommen.
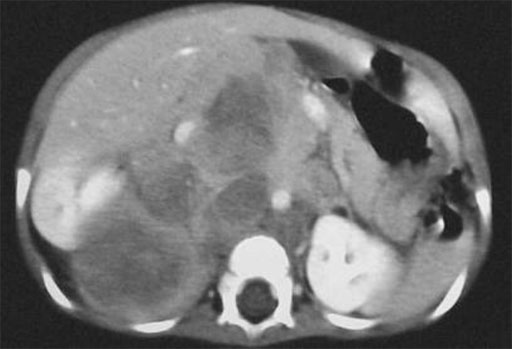
CT – Bild eines Neuroblastomes vor der re. Niere, die abgedrängt vorliegt.
Die Therapie besteht aus einer Tumorbiopsie (Diagnose und Tumottyp),, einer Knochenmarkspunktion (Typ IVs) einer Chemotherapie sowie schließlich der Resektion des Resttumors welcher bereits deutlich an Größe abgenommen haben sollte. Im Anschluss an die Tumorresektion sind weitere so genannte „Chemotherapiezyklen“ notwendig. Die operative Therapie erfordert die exakte Präparation des „Retroperitoneums“, also jenem Raum wo die Nieren und ableitenden Harnwege, die Nebennieren, Die Aorta und die große Hohlvene liegen.
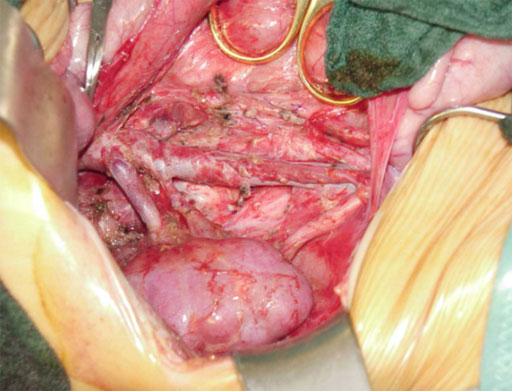
Der Tumor ist erfolgreich entfernt und die re. Niere kann wieder ihren Platz einnehmen.
Neuroblastome im 1. Lebensjahr mit radikal entferntem Tumor oder Stadium IVS überleben in über 90%: Das Vorhandensein des N-myc Onkogens in den Neuroblastomzellen ist ein prognostisch schlechtes Zeichen.
Wilms – Tumor (Nephroblastom)
Der Wilmstumor entwickelt sich aus fehlerhaft differenziertem embryonalem Nierengewebe. Als Symptome finden wir ein vorgewölbtes, oftmals einseitig ausladendes Abdomen des Kleinkindes. Man kann diesen Tumor auch gut tasten. Man findet Blut im Harn (Hämaturie) und gelegentlich Zeichen einer Obstipation. Manchmal kommt es auch zum Erbrechen. Als Untersuchung bietet sich neben dem Ultraschall die CT bzw. MR – Untersuchung an. Im Gegensatz zu dem Neuroblastom sieht man beim Nephroblastom den Zusammenhang mit dem Nierengewebe, d.h. dass der Tumor dort nicht von den gesunden Nierenanteilen abgrenzbar ist. Für die Diagnose reicht in der Regel bereits das typische CT – Bild.
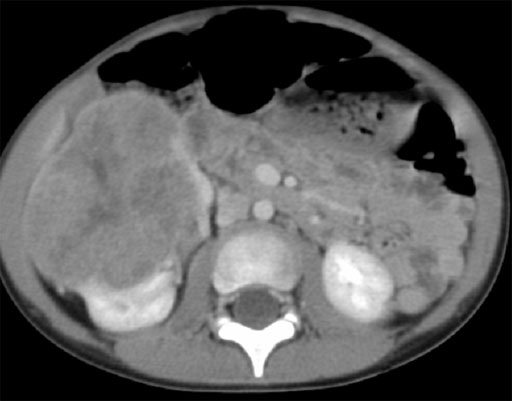
CT eines Wilms Tumors der rechten Niere.
Beim Wilmstumor unterscheidet man, je nach Größe und Ausdehnung des Tumors die Stadien I – V. Wichtig ist eine mögliche Metastasierung in Leber, Lunge und Knochenmark zu diagnostizieren. Auch Tumoreinbrüche in die untere Hohlvene kommen vor. Die Therapie besteht aus einer) einer Chemotherapie sowie der Resektion des Tumors, die in der Regel die Entfernung der Niere bedeutet. In bestimmten Fällen kann eine „nierengewebserhaltende“ Resektion vorgenommen werden. Im Anschluss an die Tumorresektion sind weitere so genannte „Chemotherapiezyklen“ notwendig. In bestimmten Fällen ergibt sich auch die Notwendigkeit einer zusätzlichen Strahlentherapie. Die Sterblichkeit der Patienten mit Wilmstumor konnte in den vergangenen Jahren erheblich gesenkt werden und liegt heute unter 20%. Wilmstumore mit günstiger Histologie haben eine Heilungsrate von 97% im Stadium I, 86% im Stadium II-III, während 57% mit ungünstiger Histologie leider nicht geholfen werden kann.
Sakrococcygeales Teratom (Steißteratom)
Dieser Tumor entsteht an der Spitze des Steißbeines bereits im Mutterleib und enthält in unterschiedlicher Ausprägung Anteile aller drei Keimblätter. Üblicherweise wächst der Tumor nach Außen, also wölbt das Gesäß vor und liegt hinter der Analöffnung. Zum Zeitpunkt der Geburt ist der Tumor in der Regel gutartig, nach 3 Monaten sind bereits bei 2/3 der Tumoren bösartige Zellnester nachweisbar. D.h. es ist eine gewisse Dringlichkeit gegeben diesen Tumor rechtzeitig zu entfernen. Mädchen sind häufiger betroffen als Knaben.
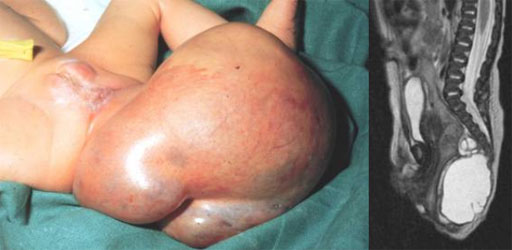
Ein großes Teratom, das sich von der Steißbeinspitze ausgehend ausgebildet hat mit zugehöriger MR – Tomographie. Man sieht den Tumor, gut abgrenzbar im Anschluss an die Steißbeinspitze.
Wesentlich zu wissen ist, dass sich diese Tumoren auch sozusagen „nach Innen“ wachsen können und hier als vorliegende Geschwulst übersehen werden können. Sie führen zurObstipation und fallweise auch zum Bettnässen, wenn der Druck auf die Harnblase zu groß ist.
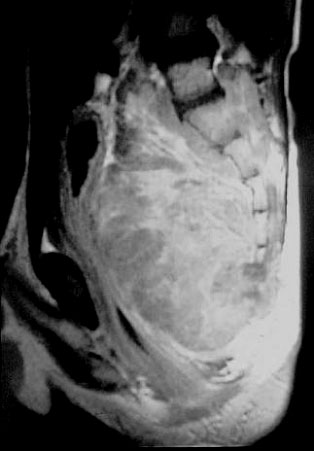
Ausgedehnter maligner Tumor vor dem Kreuzbein gelegen, der einen starken Druck auf die Harnblase und den Enddarm ausübt.




